The Human Genome Is—Finally!—Complete
The Human Genome Project left 8 percent of our DNA unexplored. Now, for the first time, those enigmatic regions have been revealed.
When the human genome was first deemed “complete” in 2000, the news was met with great international fanfare. The two rival groups vying to finish the genome first—one a large government-led consortium, the other an underdog private company—agreed to declare joint success. They shook hands at the White House. Bill Clinton presided. Tony Blair beamed in from London. “We are standing at an extraordinary moment in scientific history,” one prominent scientist declared when those genomes were published. “It’s as though we have climbed to the top of the Himalayas.”
But actually, the human genome was not complete. Neither group had reached the real summit. As even the contemporary coverage acknowledged, that version was more of a rough draft, riddled with long stretches where the DNA sequence was still fuzzy or missing. The private company soon pivoted and ended its human-genome project, though scientists with the public consortium soldiered on. In 2003, with less glitz but still plenty of headlines, the human genome was declared complete once again.
But actually, the human genome was still not complete. Even the revised draft was missing about 8 percent of the genome. These were the hardest-to-sequence regions, full of repeating letters that were simply impossible to read with the technology at the time.
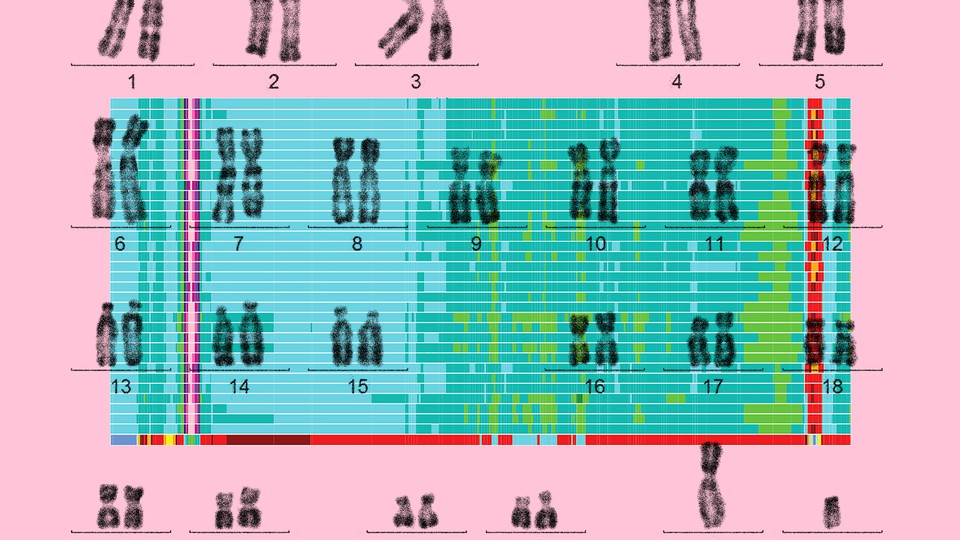
Finally, this May, a separate group of scientists quietly posted a preprint online describing what can be deemed the first truly complete human genome—a readout of all 3.055 billion letters across 23 human chromosomes. The group, led by relatively young researchers, came together on Slack from around the world to finish the task abandoned 20 years ago. There was no splashy White House announcement this time, no talk of summiting the Himalayas; the paper itself is still under review for official publication in a journal. But the lack of pomp belies what an achievement this is: To complete the human genome, these scientists had to figure out how to map its most mysterious and neglected repeating regions, which may now finally get their scientific due.
“I consider this a landmark,” says Steven Henikoff, a molecular biologist at Fred Hutchinson Cancer Research Center, who was not involved in the project. Henikoff studies one of those enigmatic, hard-to-sequence regions where previous human-genome projects had given up: centromeres, which are the slightly pinched middles of each chromosome. Chromosomes, of which humans have 23 pairs, each consist of a long, continuous stretch of DNA that can be condensed into a rod shape; the DNA at the centromere is particularly dense.
On five human chromosomes, the centromere is not in the middle but very close to one end, dividing the chromosome into one long and one very short arm. These short arms are also full of repeats that had never been entirely sequenced until now. Centromeres, short arms, and other types of repeating regions made up most of the 238 million letters the consortium ultimately added or corrected in the human genome.
The repeat-rich segments of the human genome do not usually contain genes, which is one reason they’ve long been neglected. Geneticists have focused largely on genes because their function is obvious and simple: A gene encodes a protein. (One big surprise of the earlier drafts of the human genome is how little of our DNA actually encodes proteins—only 1 percent. The role of the remaining 99 percent is becoming clearer.) Indeed, there have been hints that these repeat-rich regions also play important roles in how genes get expressed and passed on, and anomalies in them have been linked to cancer and aging. The consortium found 79 new genes hidden among the repeats too. With a map of these repeating regions finally in hand, scientists can probe more carefully their function.
The effort to finish the genome was “entirely grassroots,” says Adam Phillippy, a computational geneticist at the National Institutes of Health who co-leads the Telomere-to-Telomere (T2T) consortium that completed the genome. (Telomeres are the regions at the ends of chromosomes, so telomere to telomere means “end to end.”) Phillippy and Karen Miga, a geneticist at UC Santa Cruz, decided to create the consortium in 2018, after a call when they realized that they both harbored ambitions of finishing the human genome.
“I’m in love with repeats,” says Miga, who came to the project as a biologist trying to understand what those repeats do. Phillippy, a computer scientist by training, brought technical chops. Traditional sequencing technologies fragment DNA into small pieces, and computer algorithms have to reassemble them like puzzle pieces. The problem is that the pieces from repeating regions all look nearly the same. Now two new “long-read” sequencing technologies—called PacBio HiFi and Oxford Nanopore—allow scientists to read longer stretches of the genome. These sequencers still can’t handle chunks big enough to cross an entire centromere or a short arm, but at least the algorithms have larger puzzle pieces to assemble.
The role of centromere sequences, like many other repeating regions, is not yet fully understood, but they are most classically known as the key to cell division. When a cell divides in two, a protein spindle attaches to the centromeres, yanking the chromosomes apart to make sure that each cell gets the right number. When this goes wrong in eggs or sperm, babies can be born with chromosomal anomalies such as Down syndrome or Turner syndrome. When it goes wrong in other parts of the body, we can end up with blood cells, for example, that have too many or too few chromosomes. This is a hallmark of aging: It’s not unusual for men older than 70 to have lost the Y chromosomes in their blood cells. In one of two companion papers uploaded alongside the complete genome, the T2T consortium showed that Oxford Nanopore’s long-read technology can also be used to map where exactly the protein spindle attaches to the centromere. Examining the sequences in those regions might yield new clues to chromosomal anomalies.
The repeat-rich short arms of the chromosomes are similarly mysterious. They definitely play some role in the cellular machinery that translates genes into proteins, and knowing their sequences could shed more light on that function. Brian McStay, a biologist at the National University of Ireland at Galway, likens the complete genome to a “parts list” for chromosomes that allows scientists to try taking out the building blocks one by one. “Knowing what this parts list is, we can say, ‘This is exactly what our chromosome looks like,’” McStay says. “‘Let’s delete this and see what the impact on the function of that chromosome is.’”
As impressive as the technical feat of sequencing a complete human genome is, scientists told me that one genome is only one snapshot. Seeing how these repeating regions change over time from person to person, species to species, will be far more interesting. “What happens in cancer? What happens in development? What happens if you compare offspring to parents?” Henikoff says. The consortium proved that these repeating regions are sequenceable with the new long-read technologies. Now they can be applied to more genomes, allowing scientists to compare one with another.
Indeed, Miga says that the ultimate dream is to make every genome that scientists attempt to sequence complete from end to end, telomere to telomere. But first, the group has a more immediate goal in mind. If you wanted to fault the new genome for not being “complete,” you could point to the fact that it comprises only a single set of 23 chromosomes, whereas normal human cells have 23 pairs. To simplify the task, the group used cells from a particular type of tumor that develops from an abnormal fertilized egg and ends up with just 23 single chromosomes. The team will have to use different cells, with 23 pairs of chromosomes, to complete what is known as a “diploid” genome.
“The next major milestone would be routine diploid genomes,” says Shilpa Garg, a geneticist at the University of Copenhagen, in Denmark. Garg has used PacBio HiFi to rapidly assemble human genomes—minus some tricky regions such as the centromeres—at a rate of a few per day. That speed could help in clinical settings too, by making it easier for doctors to more regularly diagnose patients using genome sequencing. (In comparison, she says, assembling genomes from older sequencing technology takes as long as three weeks.) Truly complete genome sequencing, repeating regions and all, is getting easier and faster. Soon, another complete human genome will not be news at all.